AI-Assisted Bioinformatics
Machine learning-enabled workflows for biomarker discovery, variant prioritization, and predictive genomics.
Accelerate Drug Discovery with High-Throughput Virtual Screening and Structure-Based Molecular Docking
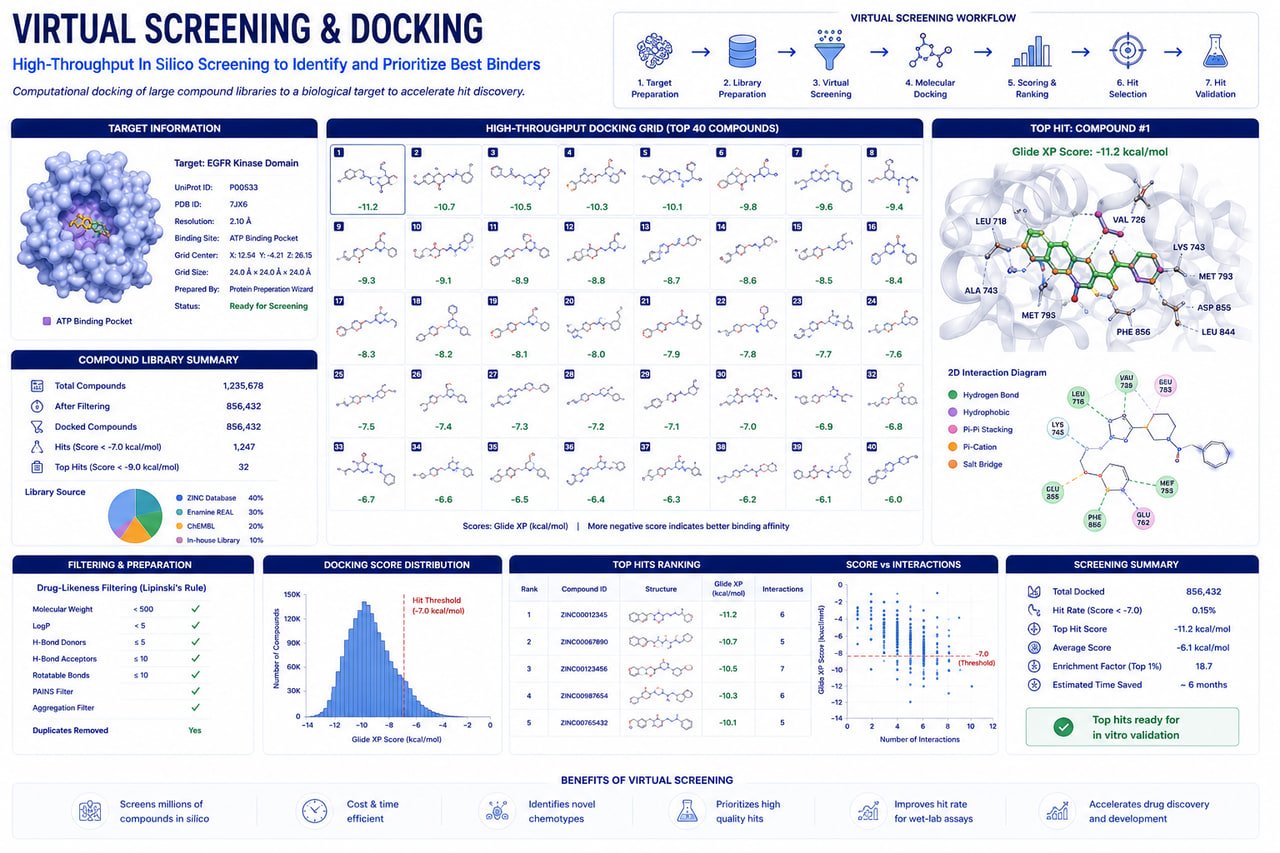
RASA Life Science Informatics provides advanced Virtual Screening & Molecular Docking services to support pharmaceutical companies, biotechnology organizations, CROs, and academic researchers in identifying promising drug candidates against validated therapeutic targets.
Our computational drug discovery platform combines structure-based virtual screening, molecular docking, pharmacophore modeling, ligand-based screening, and AI-assisted compound prioritization to rapidly evaluate large chemical libraries and identify high-confidence candidate molecules for downstream drug development.
By integrating structural bioinformatics, computational chemistry, and machine learning-driven scoring strategies, we enable efficient identification of compounds with favorable binding affinity, molecular interactions, and drug-like properties while significantly reducing experimental screening costs and timelines.
Screen thousands to millions of compounds efficiently using scalable computational infrastructure.
Support for multiple docking algorithms to improve prediction confidence and result validation.
Comprehensive evaluation of hydrogen bonds, hydrophobic contacts, electrostatic interactions, and binding site occupancy.
Pre-screening and post-screening filters to improve lead quality and reduce downstream attrition.
Containerized pipelines using Docker, Nextflow, Snakemake, and cloud-ready infrastructure.
Identification of new chemical entities against therapeutic targets.
Screening of approved and investigational compounds for new indications.
Identification and optimization of fragment hits.
Target-specific therapeutic candidate identification.
Evaluation of compounds against multiple disease-relevant targets.
Generation of high-confidence candidates for molecular dynamics simulation and lead optimization studies.
Machine learning-enabled workflows for biomarker discovery, variant prioritization, and predictive genomics.
Support for Illumina, Oxford Nanopore, PacBio HiFi, and 10x Genomics platforms.
From raw sequencing data to biological interpretation and publication-ready reports.
Deployable on AWS, Google Cloud, HPC clusters, and secure on-premise environments.
Built using Nextflow, Snakemake, Docker, and Singularity for enterprise-grade bioinformatics operations.
Get in touch with our team in Pune, India to discuss sample sizes, platform details, and custom bioinformatics pipeline configurations for your research program.
Start a Project