AI-Assisted Bioinformatics
Machine learning-enabled workflows for biomarker discovery, variant prioritization, and predictive genomics.
Enhance binding affinity, kinetic stability, and drug-like properties using GROMACS molecular dynamics, ML-driven QSAR models, and ADMET toxicity filters.
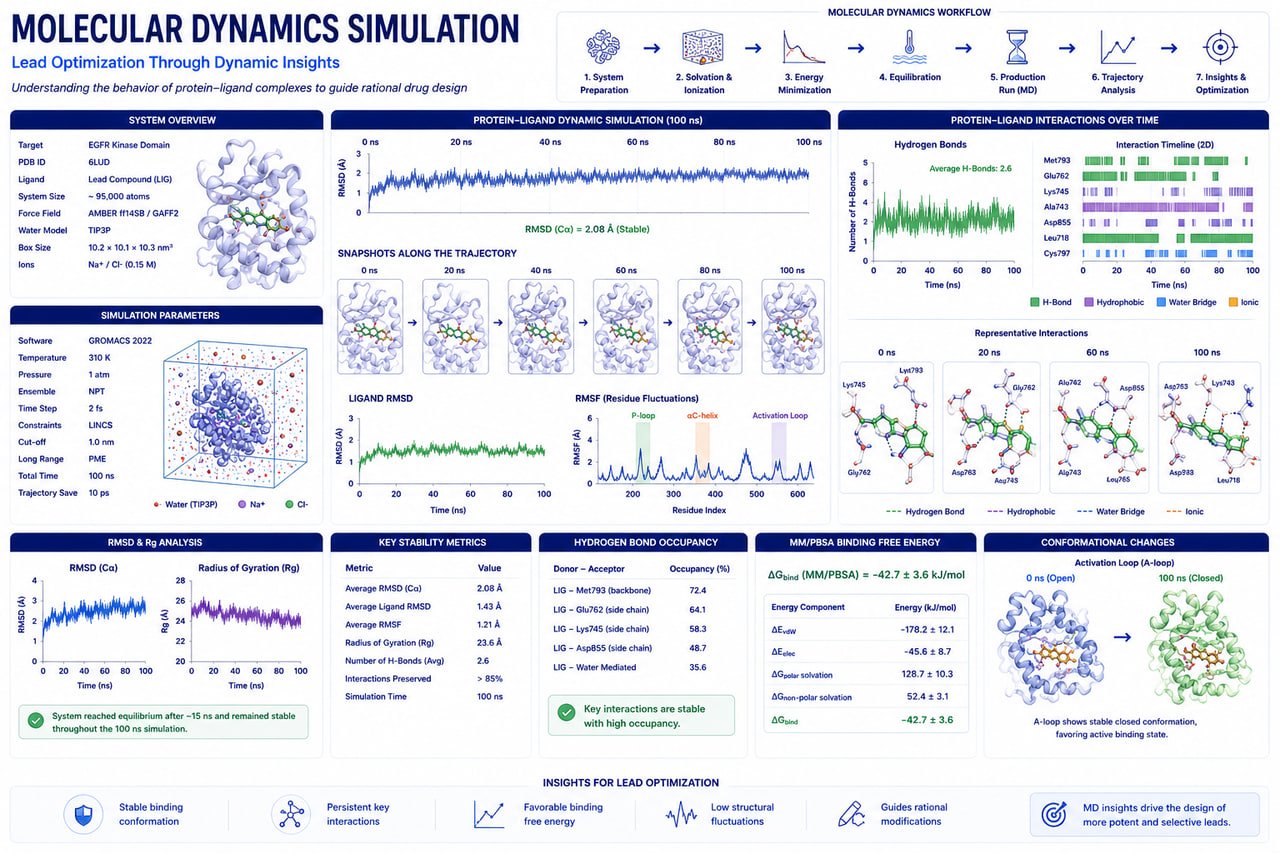
Improve target selectivity, binding affinity, and metabolic stability while minimizing off-target toxicity.
Machine learning-enabled workflows for biomarker discovery, variant prioritization, and predictive genomics.
Support for Illumina, Oxford Nanopore, PacBio HiFi, and 10x Genomics platforms.
From raw sequencing data to biological interpretation and publication-ready reports.
Deployable on AWS, Google Cloud, HPC clusters, and secure on-premise environments.
Built using Nextflow, Snakemake, Docker, and Singularity for enterprise-grade bioinformatics operations.
Get in touch with our Pune-based computational drug design experts to coordinate target screens or dynamics calculations.
Start a Project