AI-Assisted Bioinformatics
Machine learning-enabled workflows for biomarker discovery, variant prioritization, and predictive genomics.
Unlock Biomolecular Insights Through Advanced Molecular Dynamics Simulations
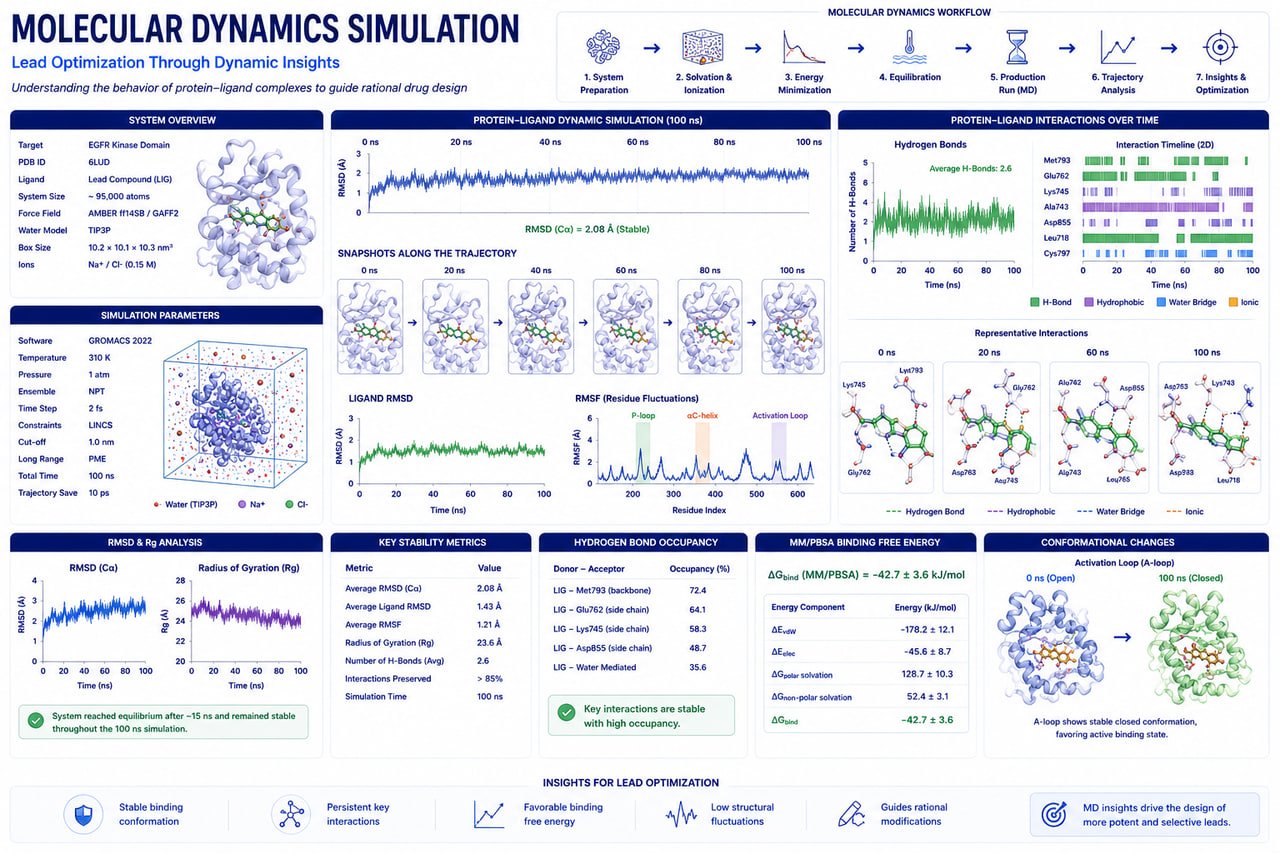
Accelerate drug discovery and biomolecular research with high-performance molecular dynamics simulations for protein structures, protein–ligand complexes, protein–protein interactions, antibodies, enzymes, and nucleic acid systems.
Evaluate protein stability, flexibility, conformational changes, and mutation effects.
Assess binding stability, interaction persistence, and drug-target dynamics.
Investigate intermolecular interactions and complex stability.
Characterize therapeutic antibody interactions and epitope recognition.
Study DNA/RNA–protein interactions and biomolecular mechanisms.
Initial stability studies — RMSD, RMSF, hydrogen bonds.
Detailed interaction analysis — PCA, DCCM, MM-PBSA.
Long-timescale conformational studies — FEL, clustering, essential dynamics.
Lead optimization and drug candidate validation.
Protein engineering and biologics development.
Computational chemistry support.
Structural biology and biomolecular research.
Mechanistic therapeutic investigations.
Machine learning-enabled workflows for biomarker discovery, variant prioritization, and predictive genomics.
Support for Illumina, Oxford Nanopore, PacBio HiFi, and 10x Genomics platforms.
From raw sequencing data to biological interpretation and publication-ready reports.
Deployable on AWS, Google Cloud, HPC clusters, and secure on-premise environments.
Built using Nextflow, Snakemake, Docker, and Singularity for enterprise-grade bioinformatics operations.
Get in touch with our team in Pune, India to discuss sample sizes, platform details, and custom bioinformatics pipeline configurations for your research program.
Start a Project