
Computer Aided Drug Designing
To Identify Potential Inhibitor/Novel Ligands for Alzheimer’s’ Disease with Computer Aided Drug Designing.
Alzheimer’s disease is one of the major causes of death today. The disease is irreversible and progresses slowly, eventually killing memory and the ability to even a simple task.
Due to the complexity of Alzheimer’s disease, the treatment involves multiple drugs. Currently, therapies aimed at maintaining mental health and slowing down the memory loss. As per the latest research, scientists are including drug therapies, physical and cognitive training, therapies for better immunity etc., as a part of clinical trials.
A cure for this disease has not been designed until now. A number of drugs are available to assist temporary relief, such as AChE Inhibitors, Memantine, Donepzil, Huperzine, Pargyline, Viloxazine, etc. In this case study, we have incorporated the known drug/compounds for finding/identifying novel potential drug/compounds for Alzheimer’s disease.
- Finding Known Inhibitors: The Two Protein Targets, as well as the known Compounds for Alzheimer’s, were selected on the basis of literature mining. The extensive literature mining was carried out using Chemical Databases & Research Articles. A total of 8 Active Compounds were finalized on the basis of their Binding Energy with respect to Target. Target Active Site prediction was performed using Literature & Online Servers such as MetaPocket.
- Designing A Pharmacophore: A pharmacophore is an essential feature of a drug responsible for its biological activity. Pharmacophores consist of structures such as Hydrogen Bond Acceptor & Donor, Positively & Negatively Charged Atoms, Aromatic Rings & Hydrophobic Regions. These Pharmacophores can be built using known Active Ligands or using Receptor Active Site Data, which are then used in Lead Identification for finding Novel Potential Inhibitors. A server such as PharmaGist can be used for Pharmacophore Designing, wherein, a complex molecule file of known compounds would be given as input & by either setting a key molecule or not a list of potential pharmacophores may be generated (Fig. 1). The list can then be further segregated on the basis of No. of Molecules Aligned, Score and Other Physio-Chemical Properties to select a final pharmacophore.
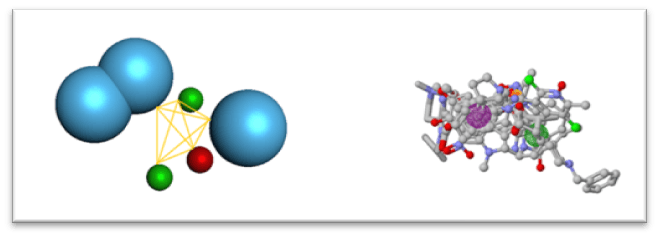
Figure 1: Pharmacophore with Its Features
- Screening Pharmacophore with ZINC Databases: A pharmacophore is screened against a database such as ZINC Human Database to get Potential Novel Compounds/Drugs. The screening is based on the features present in a pharmacophore. Filters can be applied such as No. of Conformers, Hits per Molecules, Total Hits and RMSD to further refine the screening output.
- Structure-Based Screening: The Potential Inhibitors should further be filtered using Lipinksi’s Rule of 5. Lipinski’s Rule of 5 is a rule of thumb used to find drug-likeness or to determine if a chemical compound with certain pharmacological or biological activity has properties that would make it a likely oral drug in humans. The obtained drugs would then be scrutinized for interaction with the Target Protein. A Docking study would be performed to achieve these goals. Docking is a method which is used to establish whether a & how a ligand interacts with the target. Docking would be performed using tools such as AutoDock Tools & AutoDock Vina. AutoDock Vina is used to performing multiple dockings and the top results obtained would then be analyzed using AutoDock Tools. The study is based on No. Of Bonds Formed, Binding Energy, & Other Parameters. Furthermore, Enhanced Visualization of Docking could be used for better understanding of the interaction (Fig. 2)
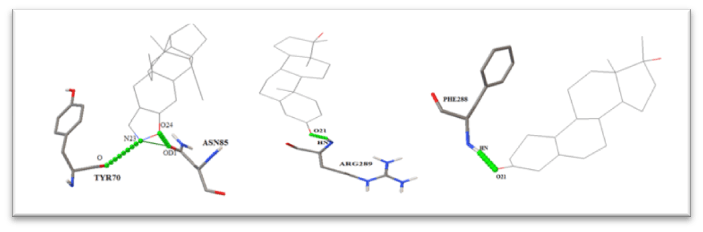
Figure 2: Interaction Images for Docking Studies
- Dynamic Studies: Simulation/Dynamic Studies could be performed for top complexes obtained after Docking. Simulation Studies are computer experiments that involve creating data by pseudorandom sampling. The key strength of simulation studies is the ability to understand the behavior of statistical methods because some parameter/s of interest are known from the process of generating the data. These studies would be analyzed for Potential Energy, Equilibration Studies and final RMSD (Fig. 3).
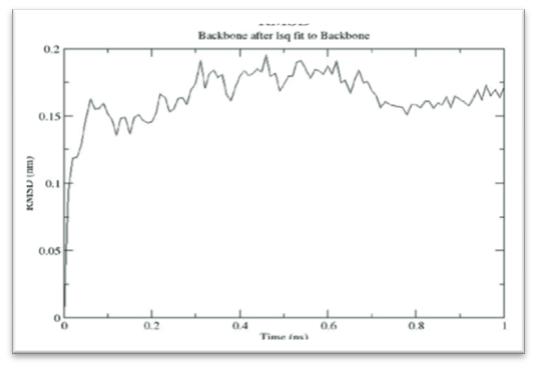
Figure 3: Graph Showing Stability of the Complex (RMSD Graph)





- Date: March 28, 2019
- Category: Computer Aided Drug Designing